郭振华研究组在简化基因组测序方法研究中取得新进展
来源:中国西南野生生物种质资源库 作者:杨国骞 2016-08-23 浏览次数:
在二代测序基础上发展起来的RAD-seq技术是一项基于全基因组酶切位点的简化基因组测序技术。由于特异性酶切位点在全基因组范围内广泛分布,通过RAD-seq能够在大多数物种内获得数万至数十万的单核苷酸多态性(single nucleotide polymorphism,SNP)标记。在此基础上,RAD-seq技术又衍生出多种简化基因组测序方法,包含GBS,ddRAD,ezRAD以及2b-RAD等。其中,ddRAD-seq通过一个稀有酶与一个常见酶相结合对基因组DNA进行双酶切,免去随机打断的过程,是一种非常有前景的简化基因组测序技术。不过,ddRAD技术虽然在建库流程上比RAD做了一定程度的简化,但仍然包括12步,实验耗时较长。因此,有必要对现有的ddRAD简化基因组测序文库构建方法进行改进,以克服其需要多次选酶、使用仪器复杂、成本偏高等缺陷,提高测序效率。
中国科学院昆明植物研究所博士研究生杨国骞在郭振华研究员与李德铢研究员指导下,与中科院海洋研究所李莉研究组一起,开发出一种被子植物中通用的双酶切简化基因组(Modified
研究以
本研究得到国家自然科学基金项目(31470322
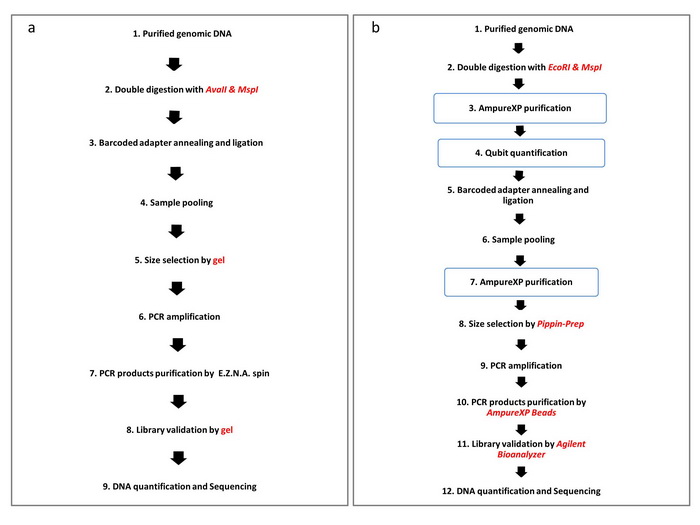
MiddRAD(a)与ddRAD(b)实验流程图
链接:http://plantmethods.biomedcentral.com/articles/10.1186/s13007-016-0139-1